



.png)

.png)
The Kitchen We Didn’t Design: Lived Experience as Advocacy

March 25, 2026
In late April, the FDA granted accelerated approval of Qalsody (Tofersen). It was the fourth therapy ever approved for the treatment of Amyotrophic Lateral Sclerosis (ALS), and first to target a genetic cause of ALS–specifically, that caused by a mutation in the SOD1 gene. This exciting FDA approval offers an opportunity to consider how we create genetic models for the ALS population not targeted by Tofersen. Below, we will briefly explore the current research that will hopefully pave the way for new effective and targeted therapies for a larger portion of the ALS population.
Mutations in the cytosolic copper-zinc superoxide dismutase 1 (SOD1) gene were first discovered to be associated with ALS in 1993 at University of Massachusetts.1 It took roughly 30 years of thorough study in the laboratory and the clinic to understand how SOD1 associated pathology could be targeted, translated into a feasible therapeutic strategy, tested in clinical trials and finally be approved by a regulatory agency.
Today, we know that SOD1 ALS accounts for roughly 2% of all ALS cases. In this form of the disease, mutations in SOD1 cause an accumulation of defective SOD1 protein in patients’ cells. These defective proteins create a toxic environment and result in motor neuron death; in jargon, we say that SOD1 mutations cause disease through a “gain-of-function” mechanism.2 This is an extremely complex process that unravels over many years (decades even); it is, however, one of the most straightforward and better understood processes in ALS pathobiology and it has been, for many years, the main model for therapeutic testing.3
Around 97% of ALS patients will not present with SOD1 pathology. These patients instead show another type of protein accumulation: TAR DNA/RNA binding protein 43 (TDP-43) proteinopathy. This is an important protein for many cell mechanisms and physiologically localizes in a specific area of the cell called the nucleus, where you will also find the DNA. In ALS patients, this protein leaves the nucleus and accumulates in a space of the cell called cytoplasm (which surrounds the nucleus), where it forms protein aggregates. In the case of TDP-43, it is less clear if these aggregates are toxic because of their presence in the cells (gain-of-function) or whether they are toxic as a result of them removing TDP-43 from the nuclei, which results in a loss of activity or “loss-of-function” of TDP-43 itself.4
TDP-43 proteinopathy has been observed in post mortem tissue of ~45% of people affected by frontotemporal dementia (FTD) and up to 57% of people affected by Alzheimer’s disease (AD).,5,6
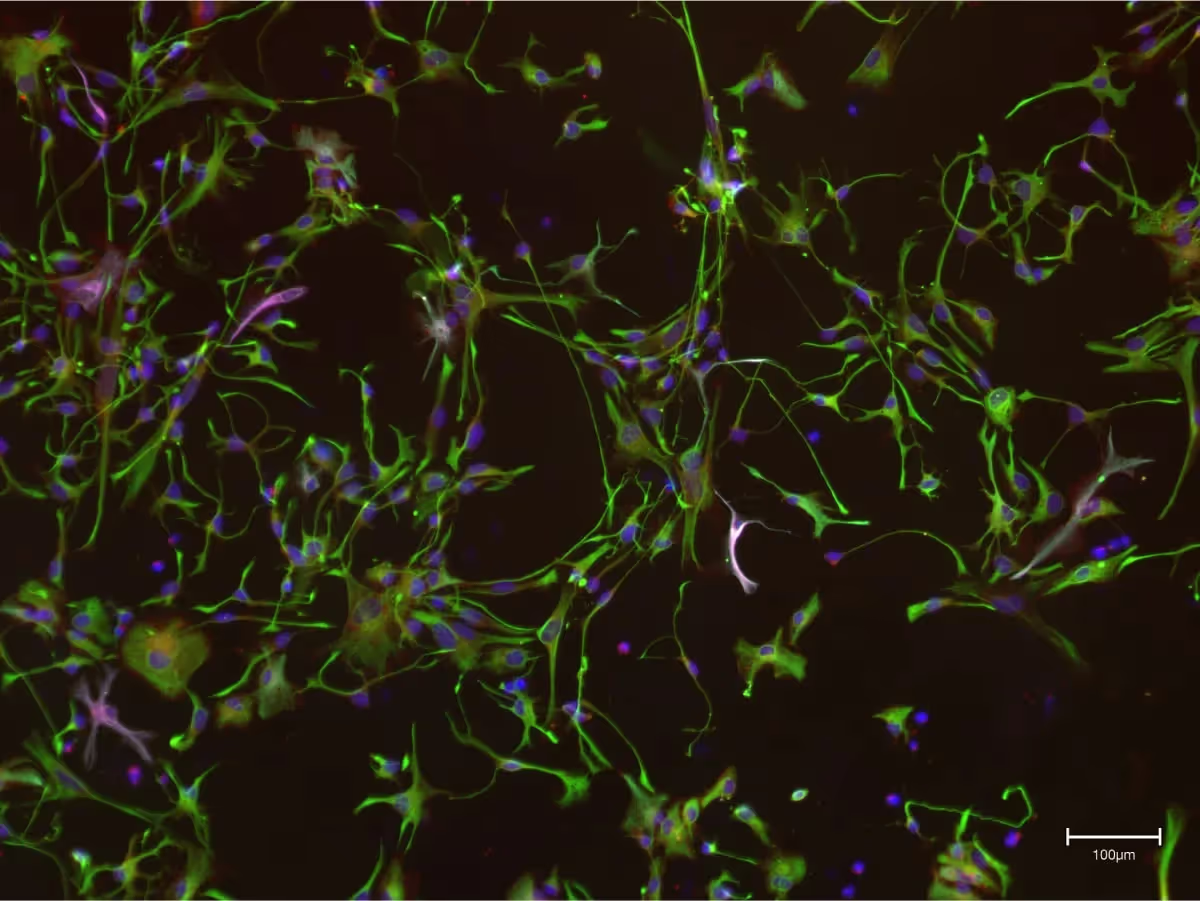
It is particularly difficult to reproduce TDP-43 pathology in vitro (i.e., in animal models) or in vivo (i.e., in a Petri dish). Most animal or cell models use either overexpression or depletion of TDP-43 in order to simulate TDP-43 gain or loss of function. Other models use some external stressors (such as starvation or chemically-induced stress) to reproduce signs of pathology.7 Only very few groups have been able to reproduce traits of TDP-43 proteinopathy in unperturbed systems.8 The therapeutic progress obtained so far has been executed using stress-induced models, which have been incredibly helpful in elucidating some key disease mechanisms. However, as techniques and knowledge progresses, accelerating therapeutic discovery requires accurate models that reproduce disease biology and pathology without inducing stress, overexpression, or depletion of TDP-43.
At Synapticure, we are using a recently published method that spontaneously reproduces most traits of TDP-43 proteinopathy in vitro, to study 3D patient-derived cell cultures.9 Said differently, this model is able to replicate what is happening in a patient’s body without having to alter or stress the cells. Our aim is to understand whether it is possible to build a library of patient-derived 3D cultures to offer a better platform for more tailored therapeutic screenings.
If you are interested in learning more about this, tune in to our upcoming seminar in collaboration with AxoSim (registration link here).
Your first consultation with a care navigator is at no cost. Give us a call at 855-255-5917 for details.
